

1. Introduction
2. Materials and Methods
Collection of seawater samples and prokaryotic abundance
DNA extraction, library construction and PCR amplification of metagenomic inserts
Identification of Antibiotic Resistance (AR) Genes
3. Results
Variability of antibiotic resistance (AR) genes
Major hosts bacterial communities of antibiotic resistance genes
4. Discussion
1. Introduction
Antibiotics are classified as synthetic, semi-synthetic, or naturally derived from microorganisms found in various ecosystems (Phillips et al. 2004; D'Costa et al. 2006). Natural habitats, including soil, animal environments, and marine ecosystems, serve as reservoirs for antibiotic resistance (AR) genes (Martínez 2008; Allen et al. 2009; Martiny et al. 2011; Forsberg et al. 2012; Hatosy and Martiny 2015). Extensive research has explored the diversity of AR genes in terrestrial and freshwater environments, such as soil (Riesenfeld et al. 2004), river sediments (Kristiansson et al. 2011), the human gut microbiome (Penders et al. 2013), fecal matter from humans (Sommer et al. 2009) and cattle (Durso et al. 2011), as well as aquaculture systems like salmon farms (Buschmann et al. 2012). Despite these studies, the presence and distribution of AR genes in the ocean's water column remain relatively underexplored (Hatosy and Martiny 2015). Wastewater sources—including sewage, pollutants, and pharmaceutical residues such as antibiotics—continuously enter marine environments. Once introduced, these contaminants undergo dilution and dispersion through ocean currents (Allison 2005; Gaw et al. 2014).
The issue of antibiotic resistance (AR) has become increasingly critical. As the use of antibiotics continues to rise, certain bacteria develop resistance mechanisms that enable them to survive in antibiotic-contaminated environments (Phillips et al. 2004; Wright 2007; Martínez 2008). In the net-cage aquafarms of Tongyeong, South Sea, Korea, approximately 71 tons of antibiotics are used annually for disease treatment and prevention (Shin 2007). Several mechanisms may explain the presence of antibiotic- resistant bacteria in marine environments (Martínez et al. 2007; Hatosy and Martiny 2015). First, resistant bacteria from terrestrial sources may be introduced into marine ecosystems through runoff. Second, the increasing use of anthropogenic antibiotics in oceans could contribute to the development of resistance in indigenous bacterial populations. Third, microbial interactions in marine environments may facilitate the acquisition of resistance genes, either through antagonistic competition or horizontal gene transfer among bacterial communities. Fourth, certain proteins, although not originally associated with resistance, may be repurposed for AR mechanisms.
Chung (2014) investigated tetracycline resistance genes in both tetracycline-resistant bacterial strains and seawater samples from the East Sea. Using a PCR-based approach with 33 primer sets including tet A, B and C genes, the study identified only the tet(C) gene in seawater samples. Phylogenetic and protein homology analyses suggested that the detected tetracycline resistance genes could be classified into novel groups of tetracycline resistance genes (Chung 2014).
However, a functional metagenomics approach provides a broader perspective on the functions of genes within microbial genomes, including AR-related genes (Pehrsson et al. 2013). Unlike traditional culture-based methods, functional metagenomics directly analyzes environmental samples such as soil, sediment, and seawater. This involves inserting DNA fragments into host cells like Escherichia coli using a vector, followed by screening for AR expression on antibiotic-containing media (Penders et al. 2013). Using functional metagenomics, Hatosy and Martiny (2015) discovered that 28% of the identified genes were already known AR genes, such as beta-lactamases and bicyclomycin resistance pumps. However, 72% of the detected genes had not been previously classified as AR- related; instead, they shared similarities with proteins such as ligases, transport pumps, oxidoreductases, and hydrolases (Hatosy and Martiny 2015). High-throughput sequencing and metagenomics analysis also revealed numerous resistance genes encoded in bacterial plasmids within fish farm sediments. These included 12 aminoglycoside resistance genes, 10 tetracycline resistance genes, 7 β-lactam resistance genes, 7 chloramphenicol resistance genes, 4 trimethoprim resistance genes, 3 glycopeptide resistance genes, 2 resistance genes for bacitracin and fluoroquinolone, a resistance gene each for macrolides, sulfonamide, and streptogramin, and 8 multidrug efflux resistance genes (Yang et al. 2013).
This study aims to investigate the distribution of antibiotic resistomes and the discovery of novel antibiotic resistance (AR) genes from the uppermost layer of the East Sea, where information on this topic remains limited.
2. Materials and Methods
Collection of seawater samples and prokaryotic abundance
Seawater samples were collected from depths of 0 m and 100 or 200 m in the East Sea (Fig. 1). Sampling was performed using 10 L Niskin bottles attached to a CTD (conductivity-temperature-depth) rosette sampler. From each depth, a total of 40–50 L of seawater was retrieved and processed immediately for prokaryotic abundance (PA) determination, DNA extraction, and storage at -20°C. PA was quantified using epifluorescence microscopy after 10–30 mL of seawater samples were fixed with 2% borate-buffered formalin, stained with DAPI (1 µg mL-1), and filtered through 0.2 μm black polycarbonate filters, following the method of Jang and Choi (2015).
DNA extraction, library construction and PCR amplification of metagenomic inserts
A total of 20 L of seawater was prefiltered using a 150 µm GF/A filter (Whatman), then collected onto a 0.22 µm, 142 mm filter (Millipore). DNA was extracted from a 0.22 µm membrane filter using phenol:chloroform:isoamyl alcohol (25:24:1) and sodium dodecyl sulfate, and quantified according to the method described by Cho and Jang (2014). At least 10 µg of DNA was fragmented to a size range of 500 to 3000 base pairs (bp) using an AIRTM DNA Fragmentation Kit (Bioo Scientific, Austin TX, USA). The fragmented DNA was processed using the End-It End Repair Kit (Epicentre, Madison, WI, USA) to generate blunt ends, following the manufacturer's instructions. Size selection of the end-repaired DNA fragments were separated by gel electrophoresis using a 1.0% low-melting agarose gel. DNA was eluted from the excised gel slices, selecting fragments between 1000 and 3000 bp, using a Gel Purification Kit (Bioneer, Daejeon, Korea).
DNA fragments were ligated into the pZE21 MCS-1 vector (Lutz and Bujard 1997) at the HincII site using a Fast-Link ligation kit (Epicentre, Madison, WI, USA) (Sommer et al. 2009), according to the manufacturer's protocol. The ligation reaction was stopped by heating at 70°C for 20 minutes.
For the transformation, the ligation mixture (4 µl) was combined with 50 µl of electrocompetent E. coli TOP10 cells (Invitrogen, Groningen, Netherlands) and transferred into a chilled 1-mm electroporation cuvette. Electroporation was carried out using a MicroPulserTM (Bio-Rad) at 1.8 kV, and the cells were immediately transferred to 1 ml of SOC medium (Sommer et al. 2009). The transformed cells were incubated at 37°C for 1 hour to recover. Afterward, the cells were then diluted 1:100 and plated onto LB agar media containing kanamycin (50 µg/ml) and incubated at 37°C.
Simultaneously, 20–200 µl of the cultured cells were plated onto LB agar plates containing combinations of kanamycin (50 µg/ml) and one of 13 different antibiotics. The following antibiotics were used: ampicillin 8 µg/ml, chloramphenicol 8 µg/ml, ciprofloxacin 8 µg/ml, erythromycin 8 µg/ml, gentamicin 8 µg/ml, nalidixic acid 4 µg/ml, Fosfomycin 8 µg/ml, rifampicin 15 µg/ml, sulfamethoxazole 16 µg/ml, polymyxin B 0.75 µg/ml, tetracycline 8 µg/ml, trimethoprim 8 µg/ml, and vancomycin 5 µg/ml. Plates were incubated at 37°C.
Template DNA for PCR amplification of the insert gene was extracted from an antibiotic-resistant clone using the boiling method, which involves suspending a single colony in sterile distilled water and heating it at 95°C for approximately 10 minutes (Englen and Kelley 2000). Colonies that failed PCR and sequences shorter than 100 bp were excluded, while the remaining 68 high-quality sequences were included in the subsequent analysis.
The crude extracts were used as the DNA template in PCR reactions, utilizing the primer pair pZE21_81_104_ 57C (5’–GAA TTC ATT AAA GAG GAG AAA GGT–3’) and pZE21_151_174rc_58C (5’–TTT CGT TTT ATT TGA TGC CTC TAG–3’) (Sommer et al. 2009). The PCR product was purified using a PCR purification kit (Bioneer, Daejeon, Korea) and subsequently sequenced on a Sanger sequencer at Macrogen, Inc. (Seoul, Korea).
Identification of Antibiotic Resistance (AR) Genes
Nucleotide sequences were compared against the non- redundant (NR) protein database in GenBank using blastx with the BLOSUM62 substitution matrix, which was developed by analyzing the frequencies of amino acid substitutions in conserved regions of proteins with less than 62% sequence identity, making it effective for detecting weak protein similarities over evolutionary distances, and an E-value cutoff of 10 (Hatosy and Martiny 2015).
3. Results
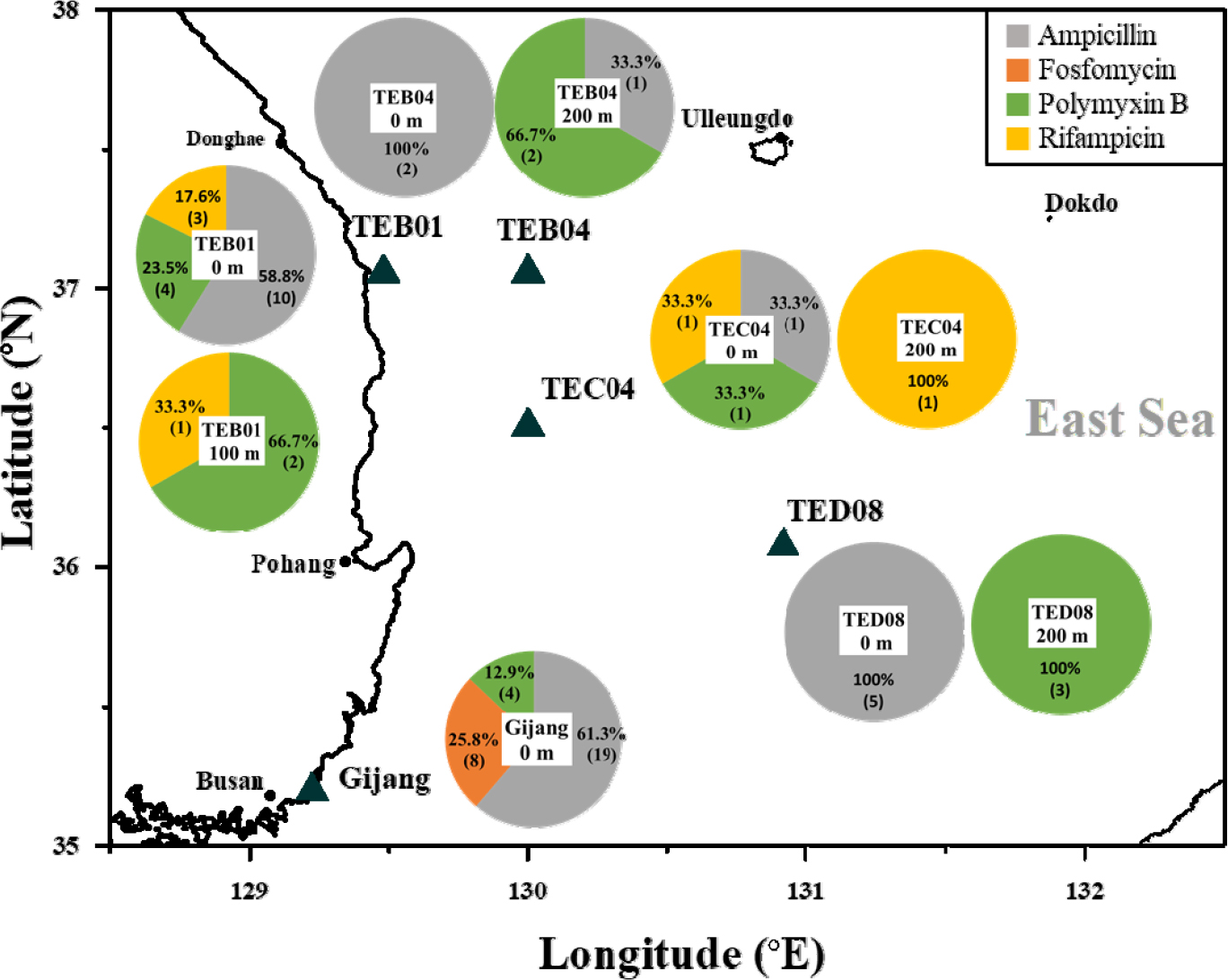
A functional metagenomic approach was employed to explore the diversity of antibiotic resistance (AR) genes in the water column (0 m, 100 m and 200 m) in the East Sea, with testing against 13 different antibiotics (see the material and methods). The total prokaryotic abundance in each sample, ranged from 0.2 to 2.2 × 106 cells per ml, measured by epifluorescence microscopy. The insert size for all libraries averaged 612 ± 262 base pairs (bp). The ratio of AR-positive clones per transformant varied from 7.6 × 10-4 to 1.1 × 10-2, relative to the transformed competent cells. Among the antibiotics tested, the majority of the cloned resistant genes were resistant to ampicillin (55.9%; 38 clones), followed by 23.5% (16 clones) resistant to polymyxin B, 11.8% (8 clones) resistant to fosfomycin, and 8.8% (6 clones) resistant to rifampicin. However, AR genes conferring resistance to 9 antibiotics - gentamicin, ciprofloxacin, chloramphenicol, erythromycin, nalidixic acid, sulfamethoxazole, trimethoprim, tetracycline, and vancomycin - were not identified in this study (Table 1).
Table 1.
Prokaryotic abundance (PA) and number of clones resistant to antibiotics across samples
None of the identified AR genes matched known AR genes in either the Antibiotic Resistance Genes Database (ARDB) or the GenBank database. Therefore, these genes were grouped by functional type based on their Enzyme Commission number such as oxidoreductase, transferase, hydrolase, lyase, isomerase, ligase, and translocase.
Variability of antibiotic resistance (AR) genes
Transferase genes, encompassing a range of functional genes (e.g., 2-phospho-L-lactate guanylyltransferase, ArnT family glycosyltransferase, carboxyl transferase domain- containing protein, DNA-directed RNA polymerase subunit beta, ketoacyl-ACP synthase III, malate synthase G, nucleotidyltransferase family protein, peptidoglycan DD- metalloendopeptidase family protein, and Uridylyltransferase [protein-PII]) were detected in all stations (Table S1).
The diversity of AR genes in surface seawater from Gijang was predominated by transferase genes (48.4%), followed by ligase genes (19.4%) (Fig. 2). Most clones were identified, conferring resistance to ampicillin, fosfomycin, and polymyxin B. Ligase genes, including coenzyme F420-0:L-glutamate ligase, were found to confer resistance to both ampicillin and fosfomycin. A higher relative abundance of clones (61.3%) was identified as resistant to ampicillin; the majority of these clones predominantly encoded transferase genes (9 clones), followed by ligases genes (2 clones). Clones conferring resistance to fosfomycin accounted for 25.8% of the total, comprising four harboring ligase genes and four harboring transferase genes. In addition, 12.9% of the clones exhibited resistance to polymyxin B, among which two carried transferase genes and two carried translocase genes (Fig. 1 and Table S1).
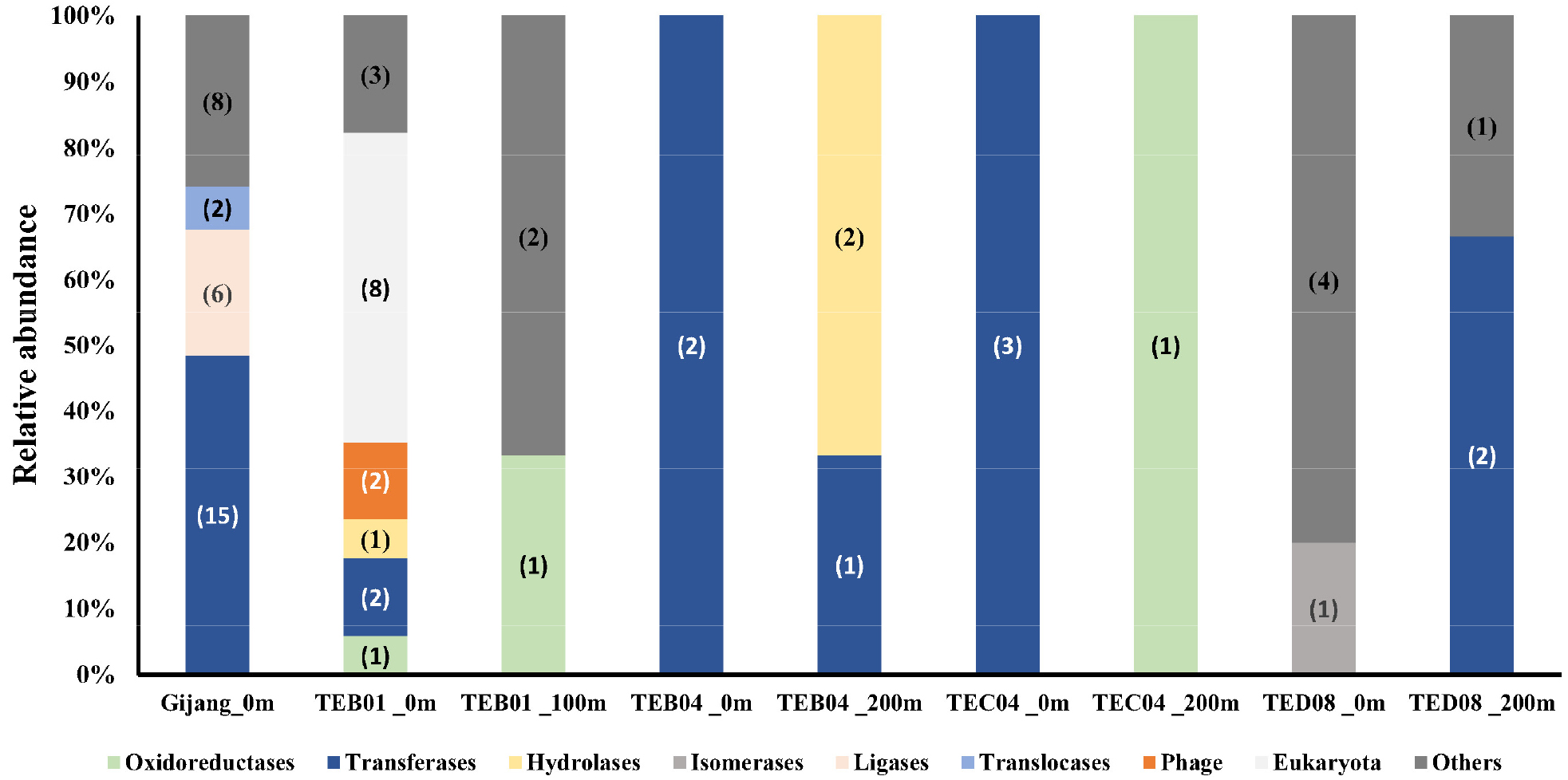
At the TEB01 station, a higher diversity of antibiotic resistance genes was detected in the TEB01-0 m sample compared to the TEB01-100 m sample. A total of eight eukaryotic genes were identified in the TEB01 0 m sample, six of which were associated with Ostreococcus sp. and two with Micromonas sp., and these genes were found to be related to resistance to ampicillin (2 clones), rifampicin (2 clones), and polymyxin B (4 clones). Two clones harboring phage genes, which were identified as tail protein genes, were detected only in the TEB01-0 m sample, and these clones exhibited resistance to ampicillin. Two clones harboring rifampicin resistance-associated oxidoreductase genes were identified, one from the TEB01 -0 m sample and the other from the TEB01-100 m sample. The gene detected at 0 m was affiliated with an acyl-CoA dehydrogenase family protein, whereas the gene from the 100 m sample was related to a 2,4-dienoyl-CoA reductase- like NADH-dependent reductase (Fig. 1 and Table S1).
At the TEB04 station, three clones harboring transferase genes, all associated with ampicillin resistance, were detected in both the 0 m and 200 m samples. In contrast, two clones associated with hydrolase genes were detected only at 200 m and were associated with resistance to polymyxin B. These hydrolase genes were specifically linked to a patatin-like phospholipase family protein (Fig. 1 and Table S1).
At the TEC04 station, four clones, harboring antibiotic resistance (AR) genes conferring resistance to ampicillin, polymyxin B, and rifampicin, were detected in both the 0 m and 200 m samples. Three clones harboring transferase genes associated with malate synthase G, conferring resistance to ampicillin, rifampicin, and polymyxin B, were detected at 0 m. A clone harboring oxidoreductase gene, associated with a GMC family oxidoreductase N-terminal domain-containing protein and conferring resistance to ampicillin, was only detected in the 200 m sample (Fig. 1 and Table S1).
At the TED08 station, eight clones, harboring antibiotic resistance (AR) genes conferring resistance to ampicillin (5 clones) and polymyxin B (3 clones), were detected in the 0 m and 200 m samples, respectively. A higher diversity of antibiotic resistance genes was detected in the TEB08-0 m sample compared to the TEB08-200 m sample. Five clones detected in the TEB08-0 m sample, conferring resistance to ampicillin, were associated with cadherin repeat domain-containing protein, D-hexose-6-phosphate mutarotase, and energy-dependent translational throttle protein EttA Three clones detected in the TEB08-200 m sample, conferring resistance to polymyxin B, were associated with carboxyl transferase domain-containing protein and elongation factor Tu (Fig. 1 and Table S1).
The distribution of protein functional types differed among antibiotics, with the exception of fosfomycin and rifampicin, which were predominantly associated with phage genes and hypothetical proteins, respectively. The distribution of protein functional types showed considerable variation in both ampicillin and polymyxin B treated samples. For ampicillin, the dominant functional types of antibiotic resistance (AR) genes were transferase and ligase, representing 39.5% (15 clones) and 5.3% (2 clones) of the total, respectively. In the case of polymyxin B, transferase, hydrolase, and translocase were the predominant functional types, accounting for 31.3% (5 clones), 12.5% (2 clones), and 12.5% (2 clones), respectively. In contrast, rifampicin treatment was associated with the identification of oxidoreductase (3 clones) and transferase (one clone), while fosfomycin treatment revealed proteins with transferase (4 clone) and ligase (4 clone) functions (Table 2).
Table 2.
Relative abundance (%) of protein functional types across antibiotic
| Functional types | Ampicillin | Rifampicin | Polymyxin B | Fosfomycin |
| Oxidoreductase | - | 50.0 (3) | - | - |
| Transferase | 39.5 (15) | 16.7 (1) | 31.3 (5) | 50.0 (4) |
| Hydrolase | 2.6 (1) | - | 12.5 (2) | - |
| Isomerase | 2.6 (1) | - | - | - |
| Ligase | 5.3 (2) | - | - | 50.0 (4) |
| Translocase | - | - | 12.5 (2) | - |
| Phage | 5.3 (2) | - | - | - |
| Eukaryota | 5.3 (2) | 33.3 (2) | 25.0 (4) | - |
| *Others | 39.5 (15) | - | 18.8 (3) | - |
*Others include an ACT domain-containing protein, a cadherin repeat domain-containing protein, an elongation factor, a hemolysin-type calcium-binding protein repeat protein, an M23 family metallopeptidase, a putative ABC transporter ATP-binding protein, a TonB-dependent receptor, a TRAP transporter fused permease subunit, and a hypothetical protein. Number of DNA fragments for each sample are shown in parentheses.
Major hosts bacterial communities of antibiotic resistance genes
The taxa of host organisms annotated from the AR genes was analyzed. Bacterial taxa were mainly composed of Alpha-proteobacteria, Gamma-proteobacteria, and Eukaryota, accounting for 42.6% (29 clones), 27.9% (19 clones), and 11.8% (8 clones), respectively. Resistance genes to ampicillin, rifampicin and polymyxin B were mostly found in Alpha- and Gamma-proteobacteria. For fosfomycin, resistance genes were only found in Gamma-proteobacteria. Alpha-proteobacteria were the predominant contributors to antibiotic resistance genes at Gijang (0 m), TEB01 (0 m and 100 m), and TEC04 (0 m). In contrast, Gamma-proteobacteria accounted for the majority of resistance genes at Gijang (0 m), TEB04 (200 m), and TED08 (0 m) (Table S1).
4. Discussion
This study identified antibiotic resistance (AR) genes associated with resistance to ampicillin, fosfomycin, polymyxin B, and rifampicin in the water column in the East Sea. None of the identified AR genes were categorized as known resistance genes. These results are consistent with those of Hatosy and Martiny (2015), who identified a diverse range of antibiotic AR genes from coastal to offshore using a functional metagenomic approach. They found that of the identified AR genes, 28% were known resistance genes, while the majority (72%) did not match known AR genes. None of the identified AR genes in the top 200 m layer matched known AR genes.
Hatosy and Martiny (2015) discussed potential mechanisms through which antibiotic-resistant bacteria might enter the ocean. Firstly, antibiotic-resistant bacteria may be transported from terrestrial environments. However, most of the AR genes in this study were derived from marine bacterial taxa. A variety of AR genes were detected in both surface water and at 200 m depths of the offshore site, located about 140 km from the coastal area. Therefore, the results cannot be fully explained by the introduction of antibiotic-resistant bacteria from terrestrial environments. Secondly, anthropogenic antibiotics used in marine environments might affect native bacteria, allowing resistance to develop in bacteria that have AR mechanisms to defend themselves. Fosfomycin, a synthetic antibiotic, had AR genes conferring resistance detected only in coastal areas, Gijang.
Antibiotic-resistant bacteria have been screened from coastal seawaters near human impact and aquaculture facilities, associated with anthropogenic influence (Dang et al. 2007; Buschmann et al. 2012; Port et al. 2012). For example, Dang et al. (2007) identified chloramphenicol resistance genes, including cat II and floR, as well as oxytetracycline (OTC) resistance genes such as tet(A), tet(B), and tet(M) in Pseudoalteromonas strains from mariculture environments in China. Buschmann et al. (2012) identified AR genes in isolated bacterial species through PCR such as Sporosarcina sp., Arthrobacter sp., and Vibrio sp. for tetA, tetB, and tetK, florfenicol, oxolinic acid, putative enzymatic inactivation genes and efflux pump genes in Chilean salmon aquaculture. Furthermore, Port et al. (2012) used metagenomics combined with pyrosequencing to detect several known AR genes for beta-lactams (BL3_imp, BL2d_oxa10, and orf11), tetracycline (tetA, tet39, tet40, and tetW), macrolides (ermB, MefA, MacB, and AcrB), bacitracin (BacA), and quaternary ammonium compounds (qacG2) in coastal areas affected by anthropogenic inputs. Additionally, Sun et al. (2014) reported that most bacterial species possess efflux pumps causing multidrug resistance, such as the RND (resistance-nodulation-division) family, ABC (ATP-binding cassette) superfamily, MFS (major facilitator superfamily), SMR (small multidrug resistance) family, and MATE (multidrug and toxic compound extrusion) family. However, this study did not detect these efflux pump systems.
The highest levels of AR clones were observed in coastal surface samples. It is possible that prokaryotes in marine environments may harbor a large reservoir of AR genes that do not currently confer resistance to antibiotics due to the absence of a promoter (Hatosy and Martiny 2015). The functional metagenomic approach is likely unable to reveal all mechanisms and functions of AR genes in marine environments. We were unable to detect resistance genes for nine antibiotics, which may be due to limitations in the heterogeneous expression of genes in the E. coli host cell (Forns et al. 1997). To better understand how these unknown AR genes confer resistance to antibiotics, further analysis is needed to elucidate their biochemical functions.
Our results indicated that Alpha- and Gamma- proteobacteria were the most dominant bacterial classes across nine seawater samples, consistent with previous findings from water samples in the western Pacific and Southern Oceans (Jang et al. 2022), Guangdong coastal areas (Xu at el. 2019), and aquaculture ponds (Fang et al. 2019).
This study aims to explore the diversity of the antibiotic resistome in the East Sea using a functional metagenomic approach. The functional metagenomic approach is more effective for discovering novel AR genes and resistance mechanisms than culture-dependent approaches, PCR, and sequencing methods. A diverse array of AR genes conferring resistance to ampicillin, fosfomycin, polymyxin B and rifampicin were found in the East Sea. Marine environments may act as potential global reservoirs for AR genes. The understanding of the mechanisms and ecological roles of AR bacteria in marine environments will be enhanced by the emergence of novel AR genes.
The sequences of antibiotic resistance (AR) genes were deposited in GenBank under accession numbers PV341027–PV341094, and can be accessed through the NCBI home page at: http://www.ncbi.nlm.nih.gov.
